Introduction
Schizomida Petrunkevitch, 1945 arachnids are commonly known as short-tailed whip scorpions, and comprise a small-bodied group (3-5 mm) with cryptic habits and a strong association with humid environments, frequently found in leaf litter and caves (Reddell and Cokendolpher 1995; Santos et al. 2013). Within this group, the genus Rowlandius Reddell and Cokendolpher 1995 includes more than 63 described species (World Schizomida Catalog 2022), with species distributed from the Caribbean islands to northeastern Brazil, occurring in tropical forests and semiarid environments (Reddell and Cokendolpher 1995; Armas 2002; Teruel et al. 2012; Santos et al. 2013; Giupponi et al. 2016). Despite their broad distribution across tropical regions, genomic resources for this group remain virtually nonexistent, limiting our understanding of their evolution, genomic architecture, and adaptive processes.
In Brazil, Rowlandius potiguar Santos, Ferreira & Buzzato, 2013 stands out as the first record of schizomid in caves in the country (Santos et al. 2013). The species was originally described from caves in the Jandaíra Formation, in northeastern Brazil, and currently is known in 29 caves in the semiarid Caatinga (Santos et al. 2013; Bento et al. 2021; Dantas et al. 2026). In the same geological formation, R. potiguar co-occurs with another congeneric species, R. tybykyra Silva, Dantas, Carvalho, Ferreira, Bento & Lima, 2026; however, the two species have never been recorded in syntopy, suggesting spatial segregation or niche differentiation (Dantas et al. 2026). Its distribution in the semiarid might be shaped by paleoclimatic changes throughout the Quaternary (Conti and Furlan 1996) and suggests a dynamic history of habitat expansion and contraction, making this species a promising model for investigating eco-evolutionary patterns in subterranean environments.
The advent of next-generation sequencing technologies has enabled the generation of draft genomes for non-model organisms, greatly expanding the ability to investigate genome structure, gene content, and evolutionary patterns across diverse taxa (Ekblom and Galindo 2011). For neglected taxa, such as schizomids, these data are essential to refine phylogenetic hypotheses and support broader comparative analyses within Arachnida. In this study, we present the first draft genome of R. potiguar. We characterize the main assembly metrics and assess genome completeness using BUSCO analyses based on Arthropoda and Metazoa datasets. By providing this resource, we contribute to advancing the understanding of Schizomida genomic biology and to increasing the representation of Neotropical subterranean invertebrates in genomic studies.
Material and methods
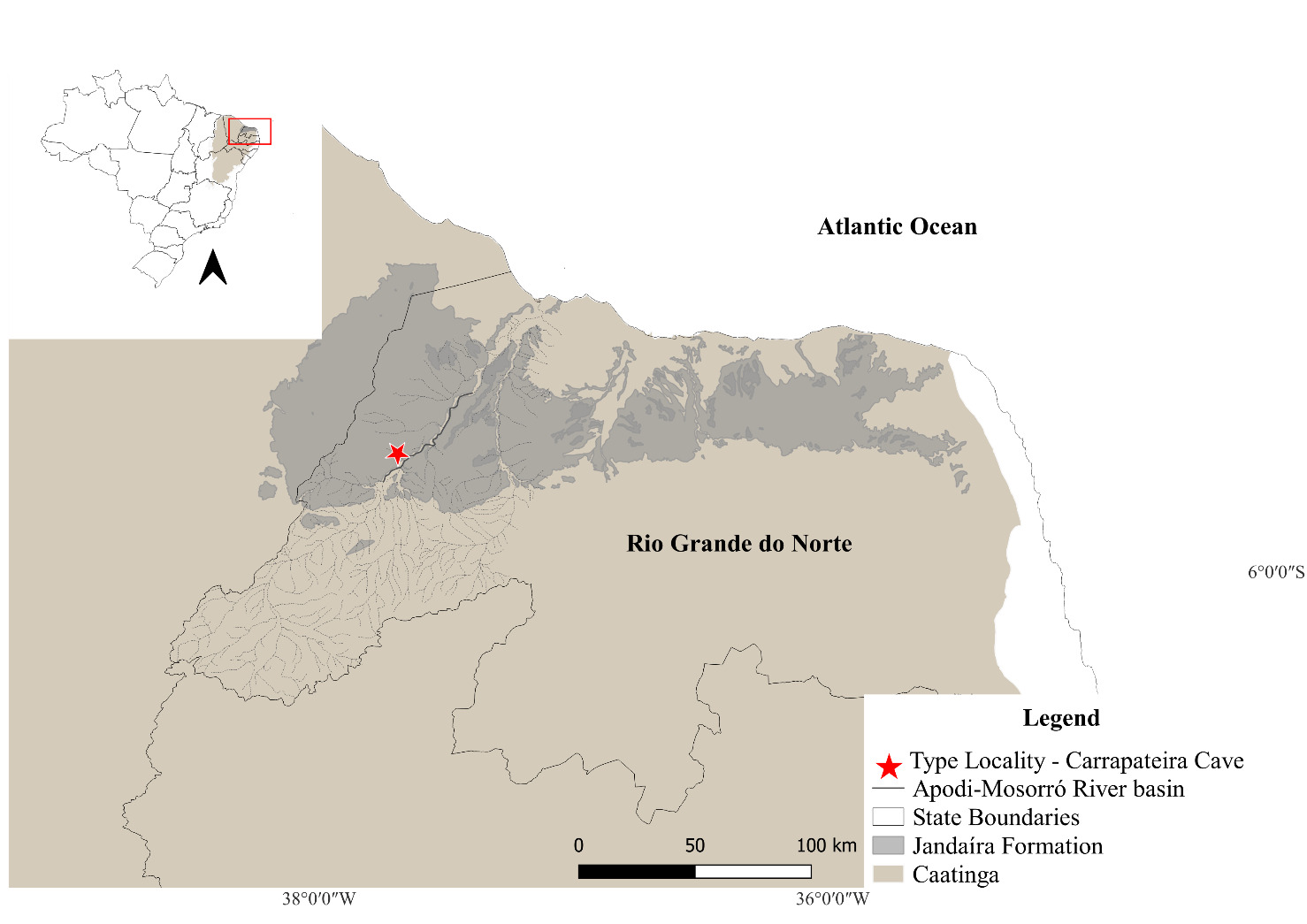
Specimens of R. potiguar used in this study were collected at its type locality (Santos et al. 2013), Carrapateira Cave (05°33′38.4″S, 37°39′50.2″W), inserted in limestones of the Jandaíra Formation, in the municipality of Felipe Guerra, Rio Grande do Norte State, northeastern Brazil (Fig. 1).
Individuals were manually collected using fine brushes through active visual searching, primarily in humid microhabitats within the cave. After collection, specimens were immediately fixed in absolute ethanol (P.A.) and stored in thermal containers during transport from the field to the laboratory, where they were subsequently kept under refrigeration. Additional specimens from the same sampling event were deposited in the Coleção de Invertebrados Subterrâneos de Lavras (ISLA) under accession numbers ISLA-118278 and ISLA-118291. Sampling was conducted under permit issued by SISBIO/ICMBio (No. 54334-11, 2023). Access to Brazilian genetic heritage was registered in the Sistema Nacional de Gestão do Patrimônio Genético e do Conhecimento Tradicional Associado (SISGEN) under registration number A06F341.
DNA was extracted using the Qiagen DNeasy genomic extraction kit, following the standard protocol. A paired-end sequencing library was then prepared using the Illumina TruSeq kit, according to the manufacturer’s instructions. Sequencing was performed on an Illumina HiSeq platform using a paired-end format of 2 × 150 bp. The resulting FASTQ files were processed to remove adapter/primer sequences as well as low-quality regions using Trimmomatic v0.33 (Bolger et al. 2014). The cleaned reads were assembled using SPAdes v2.5 (Bankevich et al. 2012), followed by a final assembly step using Zanfona (Kieras et al. 2021). Genome assembly completeness was assessed using BUSCO v5 (Simão et al. 2015) and CEGMA 2.4 (Parra et al. 2007).
Results
Genome assembly statistics and completeness
The draft genome assembly of Rowlandius potiguar resulted in a total length of approximately 1.8 Gb, distributed across 1,505,104 scaffolds and 1,510,944 contigs (Table 1). The assembly is highly fragmented, as indicated by the low scaffold and contig N50 values (3.2 kb) and the exceptionally high number of scaffolds. The L50 values (158,314 scaffolds and 160,202 contigs) further highlight the low contiguity of the assembly. The GC content was estimated at 32.5%, which is within the range reported for other arachnids (Su et al. 2025).
Genome completeness assessed using BUSCO revealed low recovery of conserved genes (Table 2). Using the Arthropoda dataset, only 20.5% of BUSCO genes were identified as complete (19.1% single-copy and 1.5% duplicated), while a high proportion was classified as fragmented (53.0%) or missing (26.5%). Similarly, analysis with the Metazoa dataset recovered 24.7% complete BUSCOs (23.4% single-copy and 1.4% duplicated), with 54.7% fragmented and 20.5% missing genes.
The predominance of fragmented BUSCOs, together with the low proportion of complete genes, reflects the highly fragmented nature of the assembly. The extremely low N50 values and the large number of scaffolds indicate limited genome contiguity, which likely compromises gene prediction accuracy and contributes to the observed patterns of incomplete BUSCO recovery (Simão et al. 2015). Additionally, approximately 25% of complete BUSCO genes contained internal stop codons, suggesting gene prediction artifacts and/or fragmentation of coding regions (Simão et al. 2015). Such characteristics are typical of draft genome assemblies generated using short-read sequencing technologies, particularly in taxa with complex genomic architecture or high repeat content (Ekblom and Wolf 2014). Despite its large genome size (~1.8 Gb), the assembly shows limited structural continuity, which may hinder downstream analyses such as gene annotation and structural genomic studies.
Nevertheless, the genome of R. potiguar represents an important genomic resource for a poorly studied subterranean arachnid lineage. Given its occurrence in cave environments, this dataset provides a foundation for future studies on genome evolution, adaptation to subterranean habitats, and the genetic basis of troglomorphic traits. Improvements through long-read sequencing or hybrid assembly approaches are expected to substantially enhance genome continuity and completeness.
Financing
Funding for sequencing was provided by Iridian Genomes (grant IRGEN_RG_2021-1345). LSM and OBD were supported by CAPES through PhD scholarships (LSM: #88887.483610/2020–00; OBD #88887.954829/2024-00). SMQL, MP, and RLF were supported by CNPq (SMQL #312066/2021–0, MP #310688/2019–1, RLF #302925/2022-8). This study was partially supported by the Speleological Compensation Commitment Agreements (TCCE 01/2018 and 01/2022), established between the Instituto Chico Mendes de Conservação da Biodiversidade (ICMBio) and Vale S.A. Financial resources were managed by the Instituto Brasileiro de DEsenvolvimento e Sustentabilidade (IABS).