Introduction
The Eurasian Woodcock (Scolopax rusticola) and the Jack Snipe (Lymnocryptes minimus) are two Charadriiformes belonging to the Scolopacidae.
The Eurasian Woodcock is one of the eight species of the Scolopax genus (Winkler, Billerman, and Lovette 2020) distributed along the Palearctic. The Eurasian Woodcock is unique as it is the only member of the wading bird family that is not dependent on wetlands and instead lives in forested areas. This is a discreet bird mainly active at night. The species is migratory and divided into two populations, the European and the Asian. The species is listed by the IUCN (2024) as at least concern and hunted in most countries.
The Jack Snipe (Lymnocryptes minimus) is the only member of the genus Lymnocryptes (Winkler, Billerman, and Lovette 2020). Jack snipe is a migrating species breeding in North Europe (Scandinavia et Siberia) and wintering in Middle East and Europe (Lepley et al. 2005; Battisti and Soldato 2018). The Jack snipe is a discret and nocturnal bird occupying wetland habitat such as petland and swamp. The species is also listed by the IUCN (2024) as at least concern and mainly hunted in Europe and Ireland.
Methods
Blood from a single wild Scolopax rusticola individual was collected during winter 2021-2022 in northeastern France. The bird belong to the Europe/South & West Europe & North Africa population (AEWA).
Blood from a single wild Lymnocryptes minimus was collected during winter 2021-2022 in northern France. The bird belong to the Northern Europe/S & W Europe & West Africa population (AEWA).
DNA preparations, sequencing and assembly analysis were performed by Genoscope (France).
Library preparation and sequencing
DNA extractions were performed on blood using either the NucleoBond HMW DNA Extraction Kit (Macherey-Nagel, Düren, Germany) or Nanobind CBB kit (Pacific Biosciences, CA, USA), following the manufacturer’s instructions. DNA was quantified by a dsDNA-specific fluorimetric quantitation method using Qubit dsDNA HS Assays (ThermoFisher Scientific, Waltham, MA). High molecular weight (HMW) gDNA quality was checked on a Femto Pulse system (Agilent, CA, USA) and the mean length of the DNA molecules was estimated to be over 90 Kb. HMW gDNA was first size-selected using Short Read Eliminator kit (Pacific Biosciences, CA, USA).
Long Read libraries were prepared using the Oxford Nanopore SQK-LSK114 kit and loaded on R10.4.1 PromethION flow cells, producing over 70 Gb of data per genome. The nanopore long reads were not cleaned and raw reads were used for genome assembly.
PCR free libraries were prepared following the Kapa Hyper Prep Kit procedure provided by Roche (Roche, Basel, Switzerland). They were sequenced on a paired-end mode with an Illumina NovaSeq 6000 instrument (Illumina, San Diego, CA, USA) using a 151 base-length read chemistry. Short Illumina reads were bioinformatically post-processed sensu Alberti et al. (2017) to filter out low-quality data.
Omni-C libraries were prepared using the Dovetail Omni-C Kit (Dovetail Genomics, Scotts Valley, CA, USA), following the OmniC Proximity Ligation Assay Non Mammalian Samples Protocol for nucleated blood (version1.0). Libraries were sequenced on the NovaSeq6000, a sequencing platform from Illumina (San Diego, CA, USA) with 2x 150 read length, and more than 170 million Omni-C reads were generated for each genome.
RNA extractions were performed using the RNeasy Plus Universal kit (Qiagen, Hilde, Germany). RNA libraries were prepared following the Illumina Stranded mRNA Prep, Ligation kit (Illumina, San Diego, CA, USA) and were sequenced with the Illumina NovaSeq6000 using a paired-end 151 base-length read chemistry. Short Illumina reads were bioinformatically post-processed sensu Alberti et al. (2017) to filter out low-quality data.
Long reads-based genome assembly
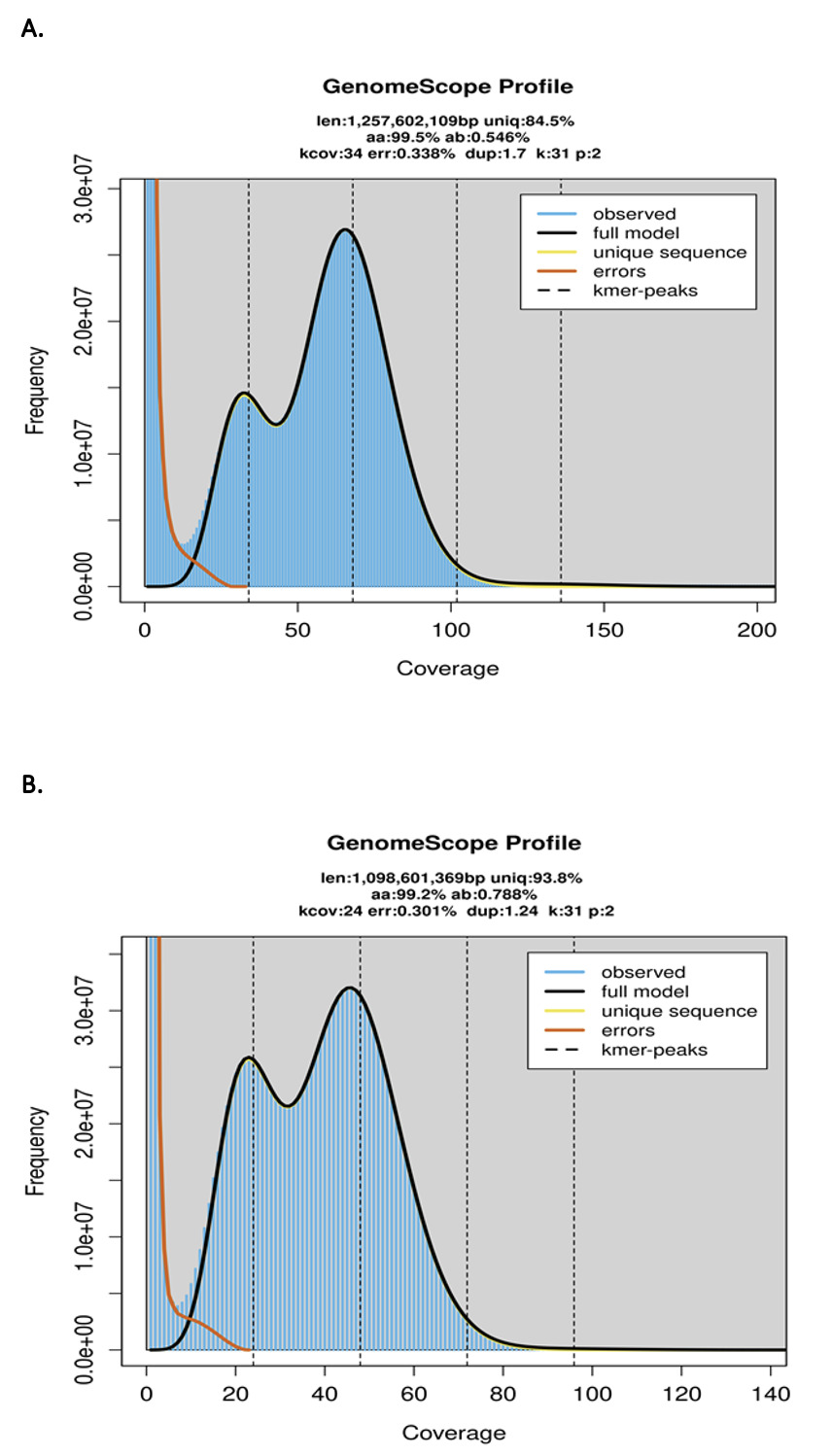
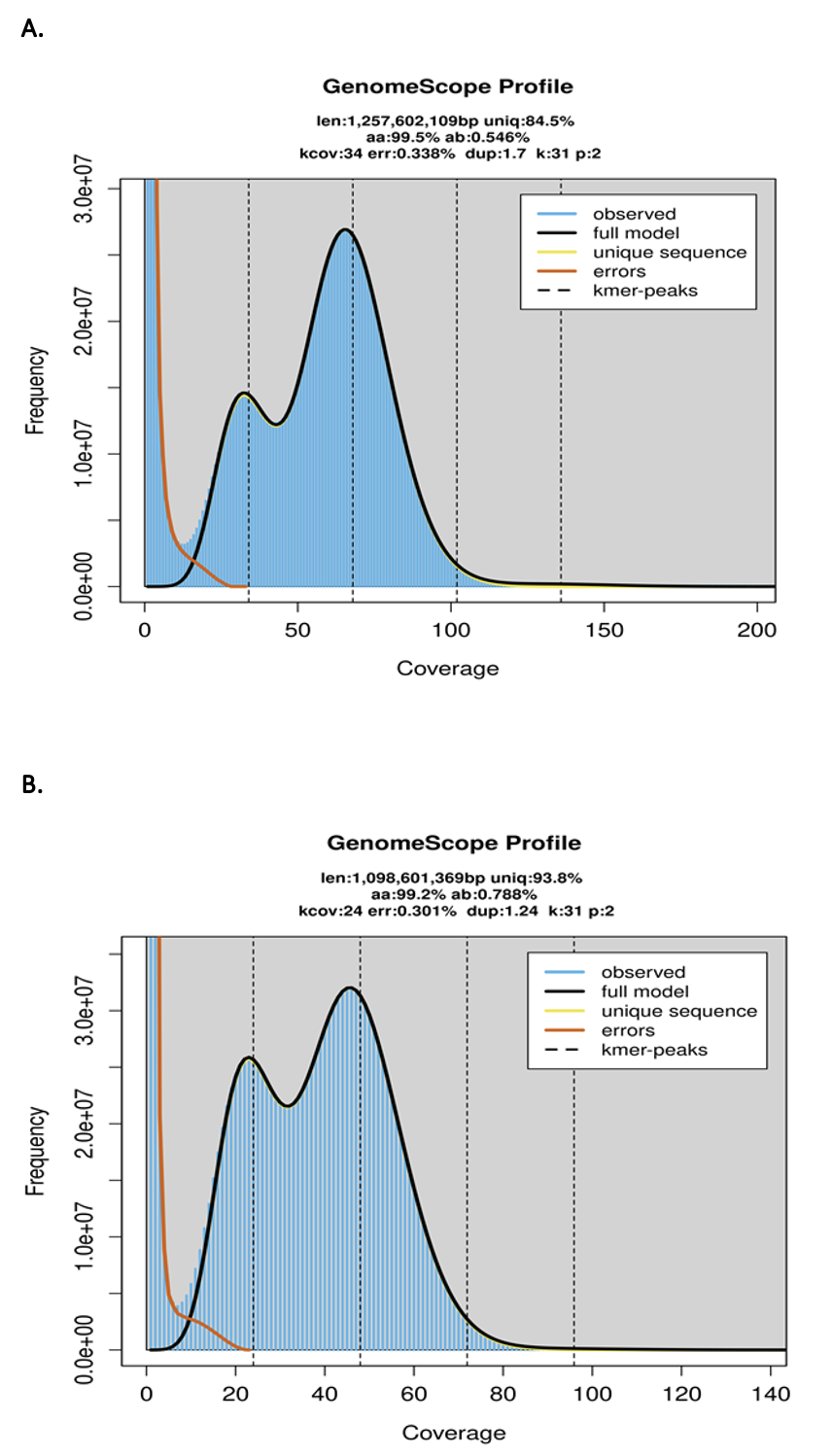
The Lymnocryptes minimus and Scolopax rusticola genome sizes were estimated using Genomescope (Ranallo-Benavidez et al. 2020), Illumina reads, and a k-mer value of 31.
To generate long-reads based genome assemblies we generated three samples of reads: (i) all read, (ii) 30X coverage of the longest reads, and (iii) 30X coverage of the filtlong (Wick 2022) highest-score reads that were used as input data for four different assemblers, Smartdenovo (Liu et al. 2021), Redbean (Ruan and Li 2020), Flye (Kolmogorov et al. 2019) and Necat. Smartdenovo was launched with -k 17 and -c 1 to generate a consensus sequence. Redbean was launched with ‘-xont -X5000 -g1200m’ and Flye with ‘-g 1200m’. The resulting assemblies were evaluated based on cumulative size and contiguity, with the Smartdenovo and all-reads combination producing the best assembly for Lymnocryptes minimus, and the Smartdenovo and filtlong reads combination for Scolopax rusticola. These two assemblies were polished using Medaka (“Medaka” 2021) with Nanopore reads and two times with Hapo-G (Aury and Istace 2021) and Illumina PCR-free reads (Table S1).
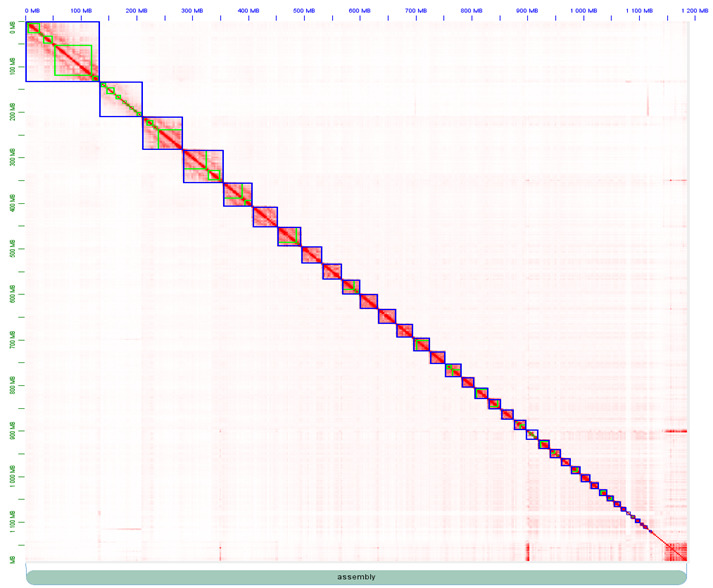
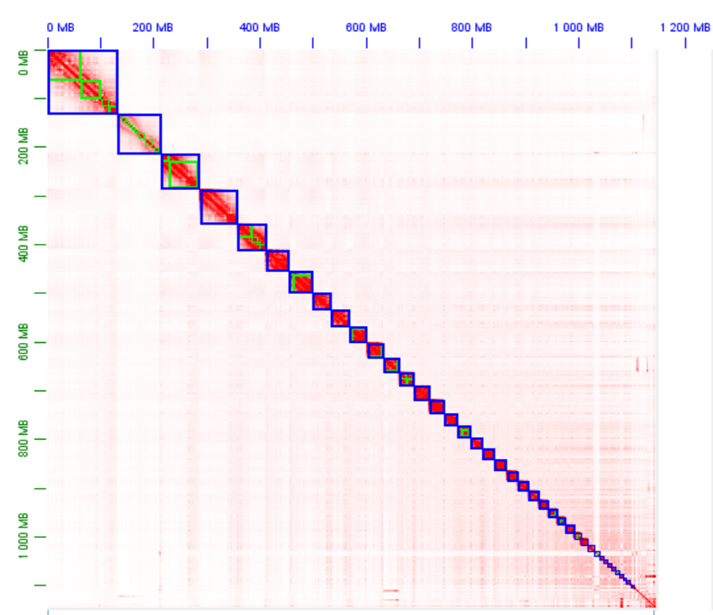
Then, both assemblies were scaffolded with Hi-C data using YaHS (Zhou, McCarthy, and Durbin 2023, 2023). The assemblies were checked for contamination and manually curated using Juicebox (Durand et al. 2016) (Figures S1 and S2).
Results and Data Availability
All raw read data and assembled genomes files are available in the GenBank database (Table 1).
The estimated genome size was 1,257Mb and 1,098Mb for L. minimus and S. rusticola (Figure S3) with a low heterozygosity rate (0.546% and 0.788%). Busco scores indicates a high-quality complete assembly with an average of 98.6% across eukaryotic databases (Table S1).
Contribution to the work
This work was part of the Limicomics project, funded by the Fédération Nationale des Chasseurs (FNC).
CB, PL conceptualized, supervised and administrated the study, organized the collect and provided the blood samples. They reviewed and edited the manuscript.
AC, KL, JMA and CC performed the DNA preparation, sequencing and assembly analysis, reviewed and edited the manuscript. (Genoscope is a platform of the Commissariat à l’Energie Atomique (CEA) and is part of the France Génomique Structure (ANR-10-INBS-09–08)).
ED and ML wrote the original draft, reviewed and edited the manuscript.
Acknowledgement
We are grateful to Christophe Urbaniak, and Benoit Balangé from the Societé de Chasse Militaire de Mourmelon. We also thank the Club International des Chasseurs de Bécassines (CICB), Jeanne Abbou and Mickaël Mourrier from the Hutte des 400 coups.